Definen subtipo independiente de la enfermedad de Castleman multicéntrica idiopática
Actualizado el 14 Nov 2022
La enfermedad multicéntrica de Castleman (EMC) comprende un grupo heterogéneo de trastornos raros que presentan síntomas generalizados como inflamación de los ganglios linfáticos, anemia, fiebre y fatiga. La enfermedad de Castleman multicéntrica idiopática (iEMC) es un tipo de enfermedad de Castleman que no está relacionada con la infección por KSHV/HHV8.
Las EMC negativas para KSHV/HHV8 sin asociación con el síndrome POEMS (polineuropatía, organomegalia, endocrinopatía, proteínas M y cambios en la piel) se denominan “idiopáticas”, es decir, enfermedades sin causa identificable, y por lo tanto se denominan “iEMC”. En la actualidad, no se han identificado manifestaciones clínicas específicas o criterios de diagnóstico para la iEMC, lo que a menudo provoca un retraso en el tratamiento.
")
Los hematopatólogos moleculares del Hospital de la Universidad de Okayama (Okayama, Japón) y sus colegas, analizaron muestras de ganglios linfáticos derivadas de 42 pacientes japoneses con compromiso de los ganglios linfáticos debido a iEMC no especificada (iEMC-NOS), que cumplieron con los criterios diagnósticos de consenso de iEMC y dieron negativo para infecciones por KSHV/HHV8. Tras un examen más detenido, el equipo clasificó a 34 de los 42 pacientes como el grupo de linfadenopatía plasmocítica idiopática (LPI) y los ocho restantes como el grupo sin LPI.
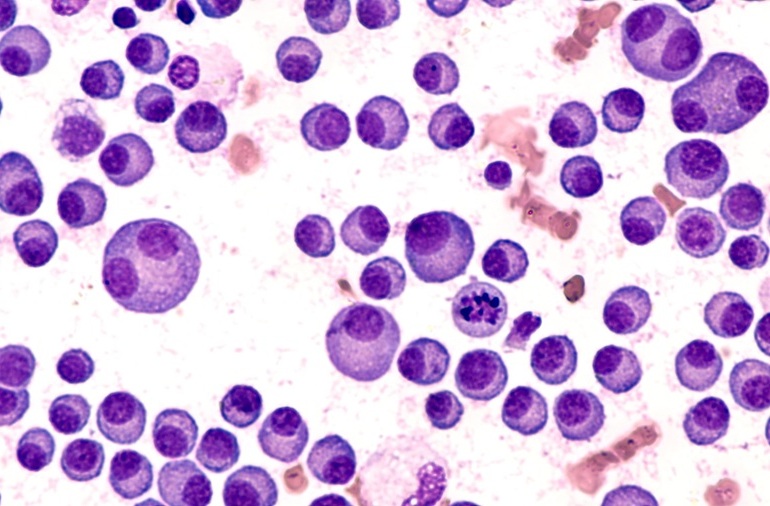
Todas las muestras de ganglios linfáticos se fijaron en formalina al 10 % y se incluyeron en parafina. Los bloques de tejido incluidos en parafina se cortaron en cortes delgados de 3 µm y se tiñeron con hematoxilina y eosina (H&E), coloración inmunohistoquímica y tinción con azul de Berlín. La coloración inmunohistoquímica se realizó con un instrumento BOND-III automatizado (Leica Biosystems, Wetzlar, Alemania) con el anticuerpo primario de HHV-8, CD138 y α-SMA. También se realizó hibridación in situ para las cadenas livianas κ y λ (Leica Biosystems).
Los investigadores informaron que, al clasificar las características histológicas como la vascularización, la plasmocitosis (proporción alta de células plasmáticas), los centros germinales (CG) atróficos e hiperplásicos, identificaron diferencias patológicas significativas entre las muestras de los grupos LPI y no LPI. El grupo de LPI demostró una mayor plasmocitosis y CG hiperplásicos en comparación con el grupo sin LPI. Por el contrario, la vascularización del grupo sin LPI fue mayor que la del grupo con LPI. Clínicamente, el grupo de LPI presentó mayor recuento de plaquetas y niveles de anticuerpos séricos (inmunoglobulina G), con menor retención de líquidos en la cavidad pleural y/o abdominal. La frecuencia de detección de autoanticuerpos específicos de la enfermedad también fue diferente entre los grupos.
Los autores concluyeron que sus resultados sugieren que la LPI es clínica y patológicamente una entidad de enfermedad uniforme y puede ser un subtipo independiente de iEMC. Se justifican estudios futuros para identificar diagnósticos, tratamientos y planes de seguimiento que sean específicos para la LPI. Dada la heterogeneidad de los casos que no son de LPI, se insta a los médicos a identificar una etiología primaria de tales casos, incluidas las enfermedades autoinmunes atípicas. Estos casos pueden beneficiarse del análisis molecular para aclarar la patología subyacente. El estudio se publicó originalmente el 7 de septiembre de 2022 en la revista International Journal of Molecular Science.
Enlaces relacionados:
Hospital de la Universidad de Okayama
Leica Biosystems