Se revela un conjunto de variantes de riesgo para la enfermedad de Hirschprung
Por el equipo editorial de LabMedica en español
Actualizado el 30 Apr 2019
La enfermedad de Hirschsprung, o aganglionosis congénita, es un trastorno del desarrollo del sistema nervioso entérico y es la causa más común de obstrucción intestinal en neonatos y lactantes, que a menudo aparece en combinación con otros síntomas de una manera que puede reflejar síndromes específicos.Actualizado el 30 Apr 2019
La enfermedad tiene una heredabilidad de más del 80%, incluidas asociaciones significativas con variantes de secuencia raras y comunes en genes relacionados con el sistema nervioso entérico, así como con síndromes monogénicos y cromosómicos. Los factores genéticos que contribuyen a una condición de desarrollo altamente heredable de la enfermedad de Hirschsprung incluyen un conjunto complejo de variantes de riesgo, que van desde polimorfismos comunes en elementos no codificantes a variantes de codificación más raras y variantes de número de copias (CNV).
.")
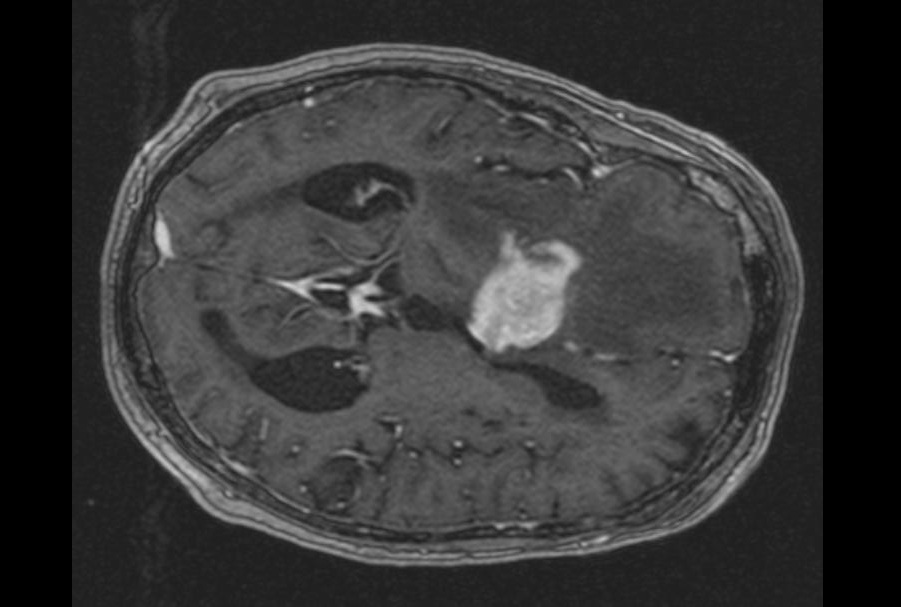
Imagen: Una histopatología de la enfermedad de Hirschsprung, caracterizada por la ausencia de células ganglionares parasimpáticas en los plexos tanto submucosos como mientéricos en el tracto gastrointestinal distal (Fotografía cortesía del Dr. Dharam Ramnani).
Científicos de varias instituciones colaboraron con los de la Universidad Johns Hopkins (Baltimore, MD, EUA) y genotipificaron y secuenciaron los exomas de 190 pacientes con enfermedad de Hirschsprung para cuantificar la carga genética en pacientes con esta afección. Las variantes de secuencia de ADN, las CNV grandes y las variantes de cariotipo en los probandos se consideraron patógenas cuando se asociaron significativamente con la enfermedad de Hirschsprung u otro trastorno del desarrollo neurológico. Estas se compararon con 627 controles no afectados, incluidos 404 participantes en el Proyecto 1000 Genomas 223, llamados “pseudo-controles”, cromosomas parentales que no se transmitieron a los niños afectados.
El equipo informó que la presencia de cinco o más variantes en cuatro elementos no codificantes definió un riesgo generalizado de enfermedad de Hirschsprung (48,4% de los pacientes y 17,1% de los controles; la relación de probabilidad fue de 4,54. Las variantes de codificación poco comunes en 24 genes que juegan un papel en el destino de las células de cresta neural entérica, siete de ellos, nuevos, también fue común (34,7% de los pacientes y 5,0% de los controles) y confirió un riesgo mucho mayor que las variantes no codificantes (relación de probabilidad, 10,02). Las CNV grandes, presentes en menos pacientes, 11,4% en comparación con el 0,2% de los controles, confirió el riesgo más alto (relación de probabilidad, 63,07). Se encontró al menos un factor de riesgo genético identificable en el 72,1% de los pacientes, y al menos el 48,4% de los pacientes tenían una deficiencia regulatoria en el gen que codifica el receptor tirosina quinasa (RET).
Los autores concluyeron que entre los pacientes en su estudio, la enfermedad de Hirschsprung surgió de variantes no codificantes comunes, variantes codificantes raras y variantes de número de copia que afectan a los genes implicados en el destino de las células de la cresta neural entérica que exacerban la susceptibilidad genética generalizada asociada con el RET. El estudio fue publicado el 11 de abril de 2019 en la revista New England Journal of Medicine.
Enlace relacionado:
Universidad Johns Hopkins

Miembro Oro
Automatic Hematology Analyzer
CF9600

Thyroid Test
Anti-Thyroid EIA Test

Repetitive Pipette
VWR® Stepper Pro