Desarrollan un análisis diagnóstico para la deficiencia inmune combinada y severa sin clasificar
Por el equipo editorial de LabMedica en español
Actualizado el 30 Jun 2020
La inmunodeficiencia combinada severa (SCID) es un grupo de trastornos genéticos hereditarios raros, y se caracteriza por una ausencia total de la función del sistema inmune, incluida la ausencia de linfocitos T, los glóbulos blancos que juegan un papel crucial en la defensa inmunológica del cuerpo. Actualizado el 30 Jun 2020
Sin el tratamiento adecuado, este trastorno es fatal durante los primeros meses de vida en la mayoría de los casos. Los programas de detección de los recién nacidos han llevado a una mayor incidencia de pacientes diagnosticados con SCID. Aunque se han identificado muchos genes causantes de SCID, los médicos pueden enfrentar a un paciente sin haber identificado algún gen anormal.
.")
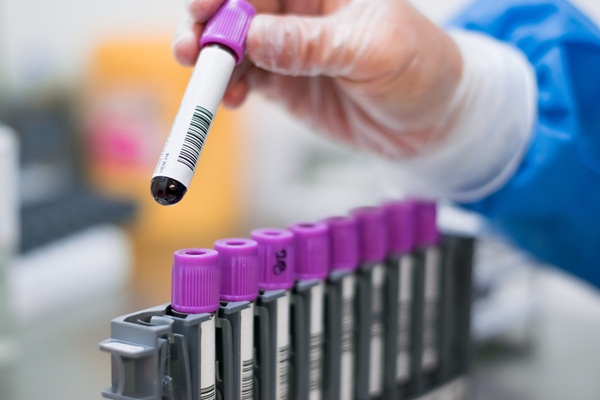
Imagen: El kit de separación celular MACS utiliza una combinación de microesferas superparamagnéticas de nanotamaño y un gradiente magnético muy alto en columnas MACS (Fotografía cortesía de Miltenyi Biotec).
Un equipo de científicos del Centro Hospitalario Universitario Sainte-Justine (Montreal, QC, Canadá) aisló un número muy pequeño de células madre de pacientes usando una cantidad limitada de sangre (3 mL a 5 mL). Se usa una prueba con un cultivo tridimensional (3D) que imita la función de un timo humano para probar esta pequeña cantidad de células, y se obtiene una respuesta en menos de cinco semanas. Si los resultados son normales, se recomienda el trasplante de timo, pero si son anormales, se prefiere un trasplante de médula ósea.
Para el cultivo en 3D, las células CD34+ se separaron de las células mononucleares por purificación con Ficoll a partir de sangre de cordón umbilical (SC) o sangre periférica (SP) fresca o congelada y descongelada, utilizando el kit MACS (Miltenyi Biotec, Bergisch Gladbach, Alemania). Las células CD34+ se mezclaron con células OP9-DLL4 cosechadas con tripsina (mezcla 1:23) para formar un comprimido celular de 2,5 µL, que se colocó en un inserto seco de cultivo de policarbonato de 25 mm. Este inserto se transfirió a una placa de 6 pozos que contenía 1,5 mL de medio. Los medios se cambiaron cada tres o cuatro días. Las células se cosecharon del inserto para realizar el análisis del clasificador de células activado por fluorescencia.
Los investigadores aplicaron el ensayo de diferenciación de células T 3D in vitro para verificar si podían diferenciar los defectos intrínsecos de diferenciación de las células madre hematopoyéticas extrínsecas (CMH) de los extrínsecos, mediante cantidades limitantes de sangre periférica de pacientes jóvenes con SCID. Como demostración de un defecto intrínseco de diferenciación, mostraron que las CMH SP-CD34+ de un paciente IL2RG/γc no podían diferenciarse en células CD34-CD7+CD1a+ doble negativo (DN), células CD4+CD8+ doble positivo (DP) o células CD3+, después de tres semanas de cultivo, aunque las células pro-T CD34+CD7+ estaban presentes en abundancia.
Por otro lado, las CMH SP-CD34+ de un paciente con SCID con deficiencia completa de RAG2 (mutación nula) avanzaron normalmente a la etapa CD34−CD7+CD1a+ DN, con escasa presencia de células CD4+CD8+ DP (0,72% versus 22,7% para el control) y sin células CD3+ después de cinco semanas de cultivo. Sin embargo, un paciente hipomorfo RAG1 adicional que se presenta con el síndrome de Omenn se podría diferenciar eficientemente hasta la etapa CD4+CD8+ DP (39,5% versus 12,36% para el control) después de cinco semanas de cultivo, pero no mostró evidencia de presencia de células CD3+.
Los autores concluyeron que presentaron una prueba de principio para un ensayo que utiliza células obtenidas de un volumen mínimo de SP para informar al médico sobre el nivel aproximado de deficiencia (células madre hematopoyéticas y células progenitoras versus defecto del timo) en el SCID no clasificado. El estudio fue publicado el 17 de junio de 2020 en la revista Blood Advances.
Enlace relacionado:
Centro Hospitalario Universitario Sainte-Justine
New
Miembro Oro
Control de preeclampsia
Acusera Pre-Eclampsia Control

Food Allergy Screening ELISA Kit
Allerquant 14G B ELISA

Repetitive Pipette
VWR® Stepper Pro